The Histone Code
Genetics, Epigenetics, and Histones
|
It is now clear that genetics won’t be able to answer all of our questions about human development and disease. These basic biological processes rely heavily on epigenetics - the ability to ‘fine-tune' the expression of specific genes.
This regulation of gene expression is essential for defining cellular identity and the dysregulation of these processes results in a variety of human diseases. Therefore, understanding these mechanisms will not only enhance our basic knowledge but will also lead to the improved detection, therapy and prognoses of several human diseases. |
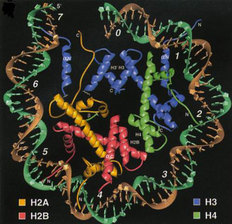
Fig. 1: The Nucleosome.
DNA is bound to an octameric complex composed of two each of histones H2A, H2B, H3, and H4. Taken from Luger et al, 1997. Nature 389:251-260. |
Histones are a group of highly evolutionarily conserved proteins that play a critical role in the proper packaging of DNA within the eukaryotic nucleus. DNA (~146 bp) along with the histones (two each of histones H2A, H2B, H3 and H4) form the fundamental repeating subunit of chromatin, known as the nucleosome (Figure 1). Since the human genome is composed of around three billion bases of DNA (3,000,000,000 bp), it is likely that there are tens of millions of nucleosomes within a single human nucleus. Because of their tight association with DNA, it has long been postulated that the histones directly participate in many different DNA-templated programs including transcription, replication, recombination and DNA repair. But there is a conundrum: if each tiny histone protein contains the same exact amino acid sequence as the other millions of histones in the nucleus, how could they possibly direct distinct and, sometimes, opposing nuclear processes (i.e. transcriptional activation versus inactivation)? One possible answer to this question has gained widespread acceptance within the last decade ?the histone code.
What is the Histone Code?
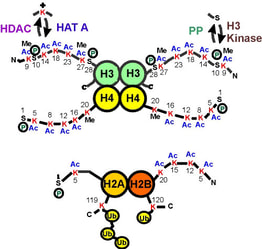
Fig. 2: A map of the histone "tails" showing modification sites.
The histone tails extend from the compact histone multimer to provide a platform for various post-translational modifications. These modifications affect the histones' ability to bind DNA and each other, which in turn affects gene expression. Taken from Strahl BD and Allis CD, 2000. Nature 403:41-45. |
The histone code hypothesis predicts that the post-translational modifications of histones, alone or in combination, function to direct specific and distinct DNA-templated programs. It has been known for over 45 years now that histones can be post-translationally modified by specific enzymes that write a histone code by adding or removing a number of different chemical modifications, including acetyl, phosphoryl and methyl groups (Figure 2). Since these modifications occur only on specific amino acid residues on specific histones in various eukaryotic organisms, these observations strongly linked the modifications involvement in nuclear processes. For example, the acetylation of key lysine residues of histone H3 and H4 by enzymes known as histone acetyltransferases (HATs) was known to play a pivotal role in transcriptional activation. |
Conversely, the removal of the acetyl groups by enzymes known as histone deacetylases (HDACs) was known to be associated with transcriptionally inactive chromatin. More recent findings demonstrate that certain histone modifications can actually block or recruit additional histone modifications. For example, methylated H4 can directly block the enzymatic activity of HATs resulting in a histone H4 that is methylated but devoid of acetylation ?this is associated with transcriptionally inactive regions. In contrast, during mitogenic stimulation, the phosphorylation of H3 can recruit HATs to acetylate H3 resulting in a histone H3 that is both phosphorylated and acetylated ?this is associated with highly transcribed regions.
Interpreting the Histone Code
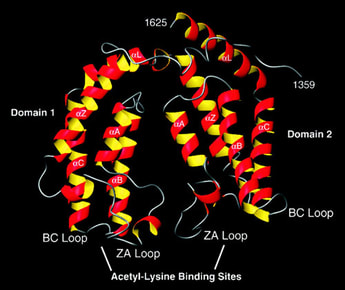
Fig. 3: The bromodomain.
The bromodomain of TAFII250, a component of the transcription initiation machinery of the cell, which specifically binds certain acetylated residues on histones. This specific binding is believed to be necessary for transcription initiation. Taken from Jacobson RH et al, 2000. Science 288 (5470): 1422-1425. |
Increasing evidence indicates that the post-translationally modified histones serve as extremely selective binding platforms for specific regulatory proteins that drive distinct nuclear processes.
How can a very simple and very small chemical modification on a comparatively huge histone make such a dramatic difference in specific nuclear functions? Recent findings indicate that certain evolutionarily conserved domains found within specific regulatory proteins possess the ability to selectively bind a certain histone modification with very high affinity. In other words, specific regulatory proteins can ‘read?the histone code to initiate DNA-templated programs. For example, the bromodomain, a conserved motif found within certain transcription factors, binds to acetylated lysine residues on histone H3 and/or H4 (Figure 3). |
|
This specific binding is believed to result in the stabilization of the transcriptional machinery at the target regions, thus, enhancing transcription of this region. In contrast, regions lacking histone acetylation are unable to effectively bind these transcription factors resulting in transcriptional inactivation of the target region. It is important to note, however, that inactive regions also contain modified histones which likely serve to maintain this inactivated state. For example, the methylation of histone H3 on lysine residue 9 (K9) was shown to occur specifically in inactivated chromatin (also known as heterochromatin). It was shown that methylated H3 K9 can recruit and bind a protein known as heterochromatin protein 1 (HP1) via its evolutionarily conserved chromodomain (Figure 4). This interaction leads to the structural formation of compacted chromatin that physically inhibits the access of the transcriptional machinery to the underlying DNA.
|
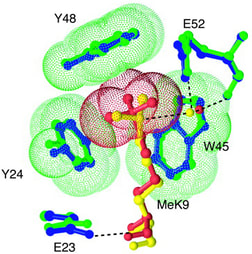
Fig. 4: The chromodomain.
A stereodiagram showing the difference in interaction between dimethylated histone H3 lysine 9 (yellow) and trimethylated H3K9 (red) as each binds to the chromodomain of HP1 (green and blue, respectively) showing the radical change the addition of a single methyl group induces in the complex. Specific binding of HP1 to the trimethylated residue is believed to be important in gene silencing. Taken from Jacobs SA and Khorasanizadeh S, 2002. Science 295(5562): 2080-2083. |
Goals of the Rice Lab
While many labs around the world are attempting to identify all the possible combinations of the histone code, our lab is asking different questions and taking different approaches to answer them?
The major goals of the Rice Lab are to discover and study the molecular pathways that establish the various histone codes and, subsequently, to elucidate their roles in human development and disease. It is clear that histone modifications define specific genomic regions (i.e. those that are transcriptionally active or inactive) and that the modifications play an essential role in all DNA-templated programs. It is also clear that histone modifications act in concert with many other nuclear factors (including the DNA, the enzymes that ‘write' the histone code and the specific regulatory proteins that ‘read' the code) to create these distinct genomic regions.
Our mission is illustrated in Figure 5:
The major goals of the Rice Lab are to discover and study the molecular pathways that establish the various histone codes and, subsequently, to elucidate their roles in human development and disease. It is clear that histone modifications define specific genomic regions (i.e. those that are transcriptionally active or inactive) and that the modifications play an essential role in all DNA-templated programs. It is also clear that histone modifications act in concert with many other nuclear factors (including the DNA, the enzymes that ‘write' the histone code and the specific regulatory proteins that ‘read' the code) to create these distinct genomic regions.
Our mission is illustrated in Figure 5:
- Goal 1: To develop and apply innovative technologies to discover the enzymatic complexes responsible for histone modifications,
- Goal 2: To identify the regulatory proteins that bind these modifications,
- Goal 3: To define the precise genomic regions targeted by histone-encoded information, and
- Goal 4: To determine the biological function(s) of each histone code.
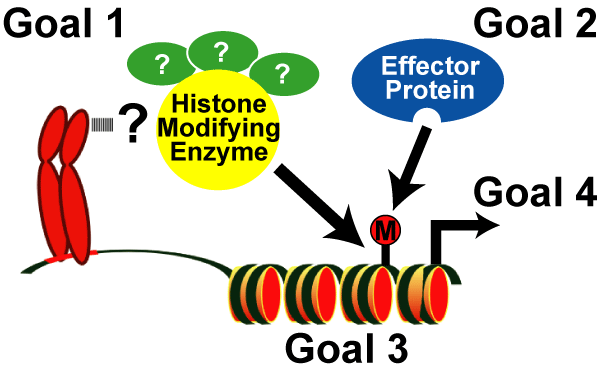
Fig. 5: Goals of the Rice Lab.
To elucidate the molecular mechanisms of the histone code and to determine their role in the epigenome. |
We propose that the histone modifications, in combination with the enzymes that ‘write' the histone code and the regulatory proteins that ‘read' the code, are critical components of eukaryotic development and differentiation pathways. Therefore, our findings will greatly enhance the fundamental understanding of the role of histone codes in normal biological pathways. Importantly, we have recently discovered that disruption of certain histone codes are associated with abnormal pathological conditions including birth defects and age-related diseases such as neurological disorders and all types of cancer. Therefore, our research will also lead to a better understanding of how the dysregulation of histone codes can contribute to various human diseases. Armed with this knowledge, our long-term goal is to translate our findings into clinical practice to improve the diagnosis, treatment and prognoses of these diseases.
|